
Pharmacokinetics: Drug Distribution
Drug distribution refers to the process by which a drug reversibly leaves the bloodstream and enters the interstitial and intracellular fluids of tissues. After a drug is absorbed into the circulation, its movement to various tissues in the body is influenced by several physicochemical and physiological factors.
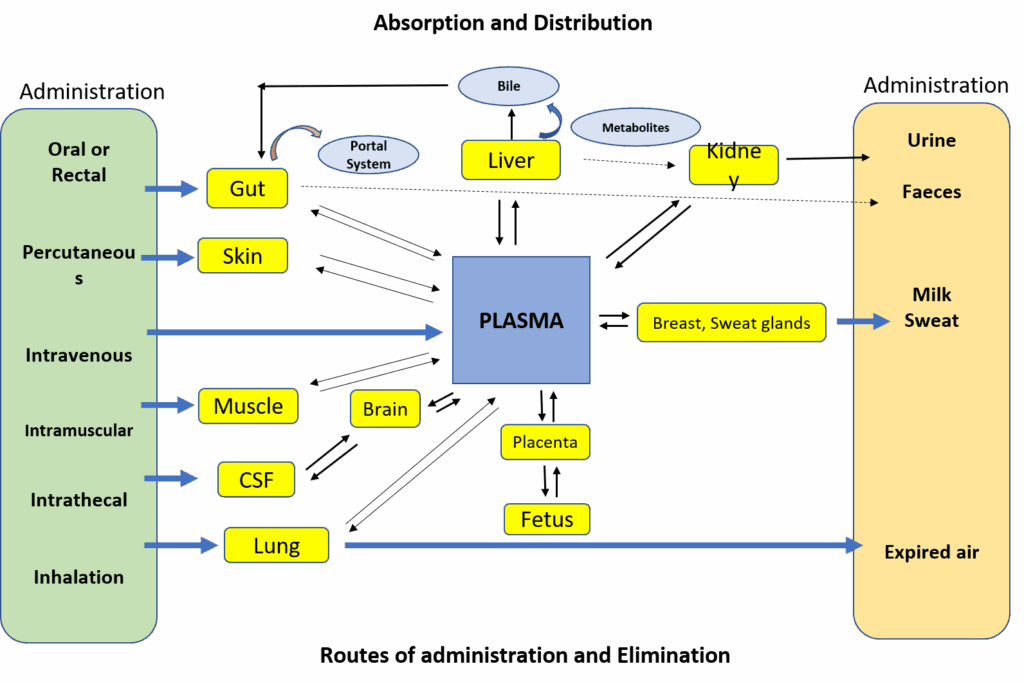
Knowing what affects how a drug spreads through the body helps us understand how it will work, how strong its effects will be, and whether it might cause any side effects.
What Affects Drug Distribution?
How quickly and how much of a drug spreads through the body depends on several things that all work together:
Fat Solubility (Lipid Solubility):
Drugs that dissolve well in fat (called lipophilic drugs) can easily pass through the body’s cell membranes. This means they can move quickly into tissues that have a lot of fat and can also cross protective barriers like the blood-brain barrier (which shields the brain) and the placenta (which separates the mother’s blood from the baby’s). On the other hand, drugs that dissolve in water (hydrophilic drugs) don’t pass through as easily.
Ionization and pKa:
Only the non-ionized form of a drug can easily diffuse across cell membranes. The degree of ionization depends on the drug’s pKa and the pH of its environment. Weak acids are better absorbed in acidic environments (e.g., stomach), while weak bases are more ionized in such settings and thus less well absorbed.
Plasma and Tissue Protein Binding:
Many drugs bind to plasma proteins like albumin (for acidic drugs) and α1-acid glycoprotein (for basic drugs). Only the unbound (free) fraction of the drug is available for distribution into tissues, metabolism, and excretion. Binding to tissue proteins may also sequester drugs in specific organs.
Regional Blood Flow:
Organs with high perfusion (e.g., liver, kidney, brain, and heart) receive more of the drug early after administration. In contrast, poorly perfused tissues (e.g., fat, skin, and bone) receive drugs more slowly.
Transport Mechanisms:
Specific transporters (e.g., P-glycoprotein, organic anion-transporting polypeptides) can facilitate or limit drug entry into cells or across barriers like the BBB and placenta.
Body Fluid Compartments
Drug distribution also depends on the body’s fluid compartments:
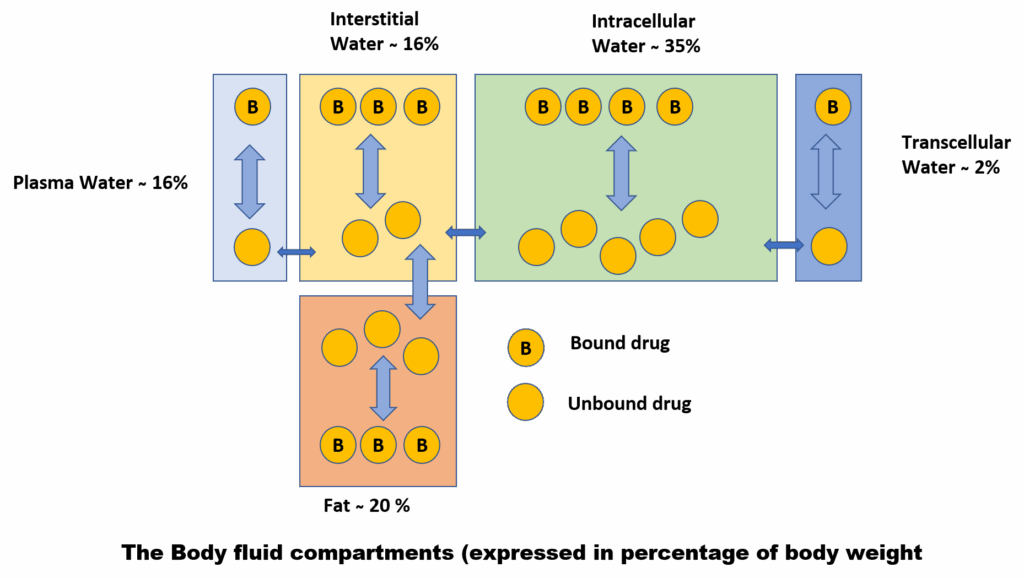
- Plasma: ~5% of body weight
- Interstitial Fluid: ~16%
- Intracellular Fluid: ~35%
- Transcellular Fluid (CSF, ocular, pleural): ~2%
- Fat: ~20% (varies with age, sex, nutritional status)
Hydrophilic drugs remain largely in the plasma and interstitial space, whereas lipophilic drugs distribute widely, including into fat.
Volume of Distribution (Vd)
Volume of distribution is a theoretical parameter that describes how extensively a drug is distributed throughout body fluids and tissues. It is defined mathematically as:
Vd = Amount of drug in the body / Plasma drug concentration
- High Vd (>0.55 L/kg or total body water): Suggests that a drug extensively distributes into tissues and may be sequestered in fat or other compartments. Examples: Digoxin, Propranolol.
- Low Vd (~0.05–0.2 L/kg): Indicates the drug remains largely in the plasma or extracellular fluid. Examples: Heparin, Gentamicin.
Vd is influenced by the same factors that affect distribution: lipid solubility, ionization, protein binding, and body composition. Disease states can also modify Vd. For instance, in congestive heart failure or liver cirrhosis, fluid retention can alter Vd, especially for hydrophilic drugs.
Redistribution
Redistribution refers to the initial rapid distribution of lipophilic drugs to highly perfused organs (like the brain), followed by their movement to less perfused tissues (e.g., fat, muscle), where they may accumulate. This phenomenon is clinically significant for drugs like thiopentone, a highly lipophilic anesthetic. After intravenous injection, thiopentone rapidly induces anesthesia by entering the brain. However, its effect is short-lived because it quickly redistributes to muscle and fat. With repeated dosing, these peripheral compartments become saturated, prolonging the drug’s action and possibly leading to toxicity.
Special Barriers to Distribution
Blood-Brain Barrier (BBB)
The BBB is a selective barrier that protects the brain from potentially harmful substances. It is composed of tight junctions between endothelial cells, supported by astrocytic foot processes. Only lipid-soluble, non-ionized drugs can cross the BBB passively.
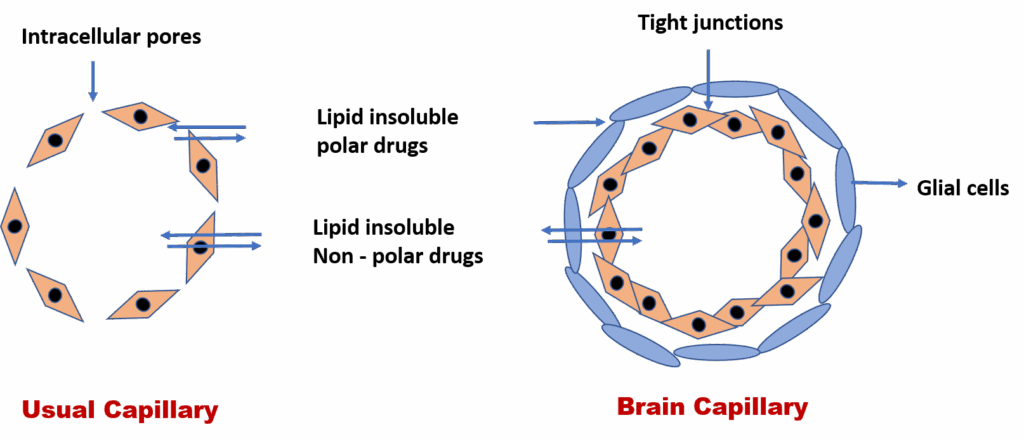
In addition, active transport systems like P-glycoprotein (P-gp) and OATP (organic anion transporting polypeptides) can actively expel drugs from the brain back into the circulation, further limiting central nervous system (CNS) exposure. This is important in drug resistance seen in CNS infections and certain cancers.
The BBB may become more permeable during inflammation, as seen in meningitis, allowing even hydrophilic drugs to reach the brain. Moreover, some areas of the brain, such as the chemoreceptor trigger zone (CTZ), lack a complete BBB. This explains how non-lipid-soluble drugs like metoclopramide can exert antiemetic effects centrally.
Placental Barrier
The placenta serves as a semi-permeable membrane between maternal and fetal circulation. Lipid-soluble, low molecular weight drugs can easily cross the placenta, exposing the fetus to potentially harmful effects. However, the placenta expresses efflux transporters like P-gp and breast cancer resistance protein (BCRP) that protect the fetus by actively pumping drugs back into the maternal circulation.
Despite these protective mechanisms, nearly all drugs can cross the placenta to some extent, especially with high maternal plasma concentrations or prolonged exposure.
Plasma Protein Binding
Drugs in the bloodstream may bind reversibly to plasma proteins. This interaction significantly affects the drug’s pharmacokinetics and pharmacodynamics:
- Albumin binds mostly acidic drugs (e.g., warfarin, phenytoin).
- α1-acid glycoprotein binds basic drugs (e.g., lidocaine, propranolol).
The bound fraction acts as a reservoir, prolonging the drug’s half-life and limiting its immediate availability for action or elimination. Only the free (unbound) drug can cross membranes, bind to receptors, and undergo metabolism or excretion.
Highly bound drugs may be displaced by other drugs with higher affinity for binding sites, temporarily increasing the free drug concentration. While this can be clinically significant in certain cases (e.g., phenytoin–valproate interaction), it is often self-limiting as redistribution and elimination restore equilibrium.
Binding percentages:
- Diazepam: ~99% bound
- Lorazepam: ~90% bound
- Flurazepam: ~10% bound
Tissue Storage
Some drugs accumulate in specific tissues due to affinity for certain cellular components:
- Tetracyclines bind calcium and are deposited in bones and teeth, potentially causing discoloration.
- Chloroquine concentrates in the retina and may cause retinopathy.
- Streptomycin accumulates in the vestibular portion of the inner ear, potentially leading to ototoxicity.
Tissue binding can result in prolonged drug action or localized toxicity, even after plasma levels decline.
Special Drug Delivery Systems
Several cutting-edge methods are used to improve medication delivery, particularly for medicines that are poorly absorbed or targeted:
- Erodible Nanoparticles: Improve absorption of oral drugs like insulin and nucleic acid-based therapies.
- Prodrugs: Inactive compounds that convert into active drugs after metabolism, aiding in solubility or targeting.
- Antibody–Drug Conjugates (ADCs): Link cytotoxic drugs to monoclonal antibodies for targeted cancer therapy.
- Liposomal Packaging: Encapsulates drugs in lipid bilayers to enhance delivery and reduce toxicity.
- Drug Implants: Provide sustained release over months to years (e.g., contraceptives, anticancer drugs).
References
Latest Editions of
- Rang H. P., Dale M. M., Ritter J. M., Flower R. J., Rang and Dale’s Pharmacology,.Churchil Livingstone Elsevier
- K.D.Tripathi. Essentials of Medical Pharmacology, JAYPEE Brothers Medical Publishers (P) Ltd, New Delhi.
- Sharma H. L., Sharma K. K., Principles of Pharmacology, Paras medical publisher